Enfermedades Hereditarias
Enfermedad Autosómica Dominante
Algunas enfermedades son heredadas en una familia de una manera dominante.
Esto significa que una persona hereda una copia normal y otra mutada de un gen y, sin embargo, la copia mutada va, a dominar sobre, o anular a la copia funcional. Esto da lugar a que el individuo esté afectado por una enfermedad genética. Qué enfermedad genética afecta a la persona en particular va a depender de las instrucciones que el gen mutado supuestamente haya dado al cuerpo.
Por ejemplo, sería el caso de enfermedades como la osteogénesis imperfecta, vinculada con genes que participan en el proceso de codificación de la proteína del colágeno básica para la estructura de los huesos. En ese caso la enfermedad se manifiesta con tan solo que tengamos mutaciones en una de las copias del gen implicado, que en el caso de la osteogénesis imperfecta en el 90 % de los casos está producida por defectos en los genes COL1A1 y COL1A2.
Algunas enfermedades genéticas dominantes afectan a la persona desde su nacimiento y, otras, sólo afectan cuando la persona a llegado a una edad adulta. Estas últimas se conocen como enfermedades de aparición tardía. Más ejemplos de enfermedades genéticas dominantes, podrían ser la poliquistosis renal dominante del adulto, la enfermedad de Huntington, la enfermedad de Caroli, el retinoblastoma, el síndrome de von Hippel-Lindau, la atrofia dentato-rubro-pallidoluysian o la distrofia miotónica de Steiner, entre otras.
* RETINOBLASTOMA

Es una enfermedad por la que se forman células malignas (cancerosas) en los tejidos de la retina.
Se presenta en forma heredable y no heredable. El tratamiento de ambas formas de retinoblastoma debe incluir orientación genética.
Los niños con antecedentes hereditarios deberán someterse a revisiones para su diagnóstico.
Un niño con retinoblastoma heredable tiene mayor riesgo de presentar un retinoblastoma trilateral.
Entre los signos y síntomas de retinoblastoma se incluyen la “pupila blanca”, y el dolor o enrojecimiento del ojo.

Para detectar (encontrar) y diagnosticar el retinoblastoma, se utilizan pruebas que examinan la retina.
Ciertos factores afectan el pronóstico (probabilidad de recuperación) y las opciones de tratamiento.
El retinoblastoma es una enfermedad por la que se forman células malignas (cancerosas) en los tejidos de la retina.

La retina es el tejido nervioso que reviste el interior de la parte posterior del ojo. Detecta la luz y envía imágenes al cerebro a través medio del nervio óptico.
A pesar de que el retinoblastoma se puede presentar a cualquier edad, aparece con mayor frecuencia en niños menores de 2 años. El cáncer puede estar en un solo ojo (unilateral) o en ambos ojos (bilateral). El retinoblastoma normalmente no se suele diseminar a los tejidos cercanos o a otras partes del cuerpo.
Se considera que un niño tiene una forma heredable de retinoblastoma cuando se produce una de las siguientes situaciones:
* Antecedentes familiares de retinoblastoma.
* Existe cierta mutación (cambio) en el gen RB1. La mutación en el gen RB1 puede pasar de uno de los padres al niño o se puede presentar en el óvulo o espermatozoide antes o poco después de la fecundación.
* Hay más de un tumor en el ojo o hay un tumor en ambos ojos.
* Se presenta un tumor en un ojo y el niño es menor de 1 año.
Después de que el retinoblastoma heredable se diagnosticó y trató, algunas veces se forman nuevos tumores durante algunos años. Habitualmente se realizan exámenes del ojo para verificar si hay nuevos tumores cada 2 a 4 meses durante por lo menos 28 meses.
El retinoblastoma heredable también aumenta el riesgo de que el niño presente otros tipos de cáncer, como cáncer de pulmón, cáncer de vejiga o melanoma en los años posteriores. Son importantes los exámenes de seguimiento regulares.
Entre los signos y síntomas de retinoblastoma se incluyen la “pupila blanca”, y el dolor o enrojecimiento del ojo.
El retinoblastoma u otras afecciones pueden causar estos y otros signos y síntomas. Consulte con un médico si su hijo presenta algo de lo siguiente:
- La pupila del ojo tiene aspecto blanco en lugar de rojo con el brillo de la luz.
- Los ojos parecen mirar en distintas direcciones (ojo vago).
- Dolor o enrojecimiento en el ojo.
- Infección alrededor del ojo.
- El globo ocular es más grande que lo normal.
- La parte del ojo con color y la pupila se ven borrosas.
Para detectar (encontrar) y diagnosticar el retinoblastoma, se utilizan pruebas que examinan la retina.
Se utilizan las pruebas y los procedimientos siguientes:
Examen físico y antecedentes: examen del cuerpo para verificar signos generales de salud, incluso el control de signos de enfermedad, como tumores o todo lo que tenga apariencia inusual. Se tomará también los antecedentes del paciente en relación con los hábitos de salud, las enfermedades y los tratamientos anteriores. El médico preguntará si hay antecedentes familiares de retinoblastoma.
Examen ocular con pupila dilatada: examen del ojo para el que se dilata (se abre más) la pupila con gotas medicinales para los ojos que permiten al médico mirar la retina y la pupila a través de una lente. Se examina con una luz el interior del ojo, incluso la retina y el nervio óptico. Según la edad del niño, este examen se puede hacer con anestesia.
Hay varios tipos de exámenes de la vista que se realizan con la pupila dilatada:
Oftalmoscopía: examen del interior de la parte posterior del ojo con una lupa pequeña y una luz para revisar la retina y el nervio óptico.
Biomicroscopia con lámpara de hendidura : examen del interior del ojo con un haz de luz fuerte y un microscopio para revisar la retina, el nervio óptico y otras partes del ojo.
Angiografía con fluoresceína: procedimiento para revisar los vasos sanguíneos y el flujo de sangre en el interior del ojo. Se inyecta en un vaso sanguíneo del brazo un tinte fluorescente anaranjado que se llama fluoresceína y que entra en el torrente sanguíneo. A medida en que el tinte recorre los vasos sanguíneos del ojo, una cámara especial toma fotos de la retina y la coroides para identificar cuáles son los vasos sanguíneos obstruidos o rotos.
Prueba del gen RB1: examen de laboratorio usado para examinar una muestra de sangre o tejido y determinar si hay un cambio en el gen RB1.
Examen del ojo con ecografía : procedimiento para el que se hacen rebotar ondas sonoras de alta energía (ultrasónicas) en los tejidos internos del ojo para producir ecos. Se utilizan gotas para adormecer los ojos, y se coloca cuidadosamente una sonda pequeña en la superficie del ojo que envía y recibe ondas sonoras. Los ecos forman una imagen del interior del ojo y se mide la distancia entre la córnea y la retina. La imagen, que se llama ecograma aparece en la pantalla del monitor de la ecografía. La imagen se puede imprimir para observar después.
Imágenes por resonancia magnética (IRM): procedimiento en el que se utiliza un imán, ondas de radio y una computadora para crear imágenes detalladas de áreas internas del cuerpo, como el ojo. Este procedimiento se también se llama imágenes por resonancia magnética nuclear (IRMN).
Exploración por TC (exploración por TAC): procedimiento mediante el cual se toma una serie de imágenes detalladas del interior del cuerpo, como el ojo, desde ángulos diferentes. Las imágenes son creadas por una computadora conectada a una máquina de rayos X. Se inyecta un tinte en una vena o se ingiere para que los órganos o los tejidos se destaquen más claramente. Este procedimiento también se llama tomografía computarizada, tomografía computadorizada o tomografía axial computarizada.

* Osteogénesis imperfecta

También es conocida como enfermedad de los huesos de cristal, es una enfermedad que debilita los huesos y hace que se rompan con facilidad sin ninguna causa aparente. La osteogénesis imperfecta puede causar también otros problemas como músculos débiles, dientes quebradizos y sordera.
La osteogénesis imperfecta es causada por una alteración genética en los huesos. Los genes contienen toda la información sobre nuestra herencia familiar y son la causa del parecido entre los miembros de una misma familia. Todos tenemos dos copias de cada gen: una por cada padre.
Cada uno de los genes que originan la osteogénesis imperfecta están relacionados de alguna manera con la producción de colágeno en el cuerpo. El colágeno es el material que ayuda a mantener los huesos fuertes. Cuando estos genes dejan de funcionar como deben, no se produce suficiente colágeno o el colágeno no funciona adecuadamente. Esto hace que los huesos se debiliten y se rompan con facilidad.
La mayoría de los niños heredan el gen que no funciona adecuadamente de uno de los padres. Algunos lo heredan de ambos padres. En algunos casos, ninguno de los padres le transmite el gen a su hijo. Simplemente el gen falla poco después del momento de la concepción.
Síntomas

Las personas que padecen de osteogénesis imperfecta tienen huesos quebradizos. La osteogénesis imperfecta puede variar de leve a grave y los síntomas son diferentes en cada persona. Algunos de los síntomas que podrían manifestar las personas que padecen osteogénesis imperfecta son:
malformaciones de los huesos, baja estatura y cuerpo pequeño, articulaciones laxas (flojas), músculos débiles, escleróticas (la parte blanca del ojo) azules, moradas o grises, cara triangular, caja torácica en forma de barril, columna vertebral curva, dientes quebradizos, sordera (generalmente comienza a los 20 ó 30 años de edad), problemas respiratorios, falta de colágeno.

Diagnóstico
No hay una prueba específica que permita diagnosticar la osteogénesis imperfecta. Para hacer el diagnóstico, los médicos utilizan:
- Antecedentes médicos familiares
- Antecedentes médicos del paciente
- Examen físico
- Radiografías
Tratamiento
Aunque no hay una cura para la osteogénesis imperfecta, se pueden aliviar los síntomas. Los tratamientos pueden incluir: tratamiento de fracturas, tratamiento de dientes quebradizos, medicamentos para aliviar el dolor, fisioterapia, el uso de sillas de rueda, corsés ortopédicos y otros aparatos,
cirugía.
Enfermedad Autosómica Recesiva
Fibrosis Quística

Es una enfermedad genética hereditaria que produce una alteración en la secreción de mucosidades, afectando a muchos sistemas del cuerpo, especialmente al sistema respiratorio y al digestivo.
El moco es una sustancia que lubrica y protege la superficie del tracto respiratorio, el sistema digestivo, el sistema reproductor y muchos otros órganos y tejidos. La fibrosis quística produce un espesamiento y aumento de la viscosidad del moco que se acumula y como consecuencia se dificulta la respiración, y se producen infecciones e irritaciones pulmonares.
También puede impedir el paso al intestino de las enzimas digestivas producidas en el páncreas que son necesarias para una correcta digestión de los alimentos, produciendo diarrea, malnutrición y pérdida de peso. Debido al bloqueo de los conductos del sistema reproductor, la fibrosis quística también puede ser causa de esterilidad.
La fibrosis quística no tiene cura y solía ser una enfermedad fatal en la infancia. Pero gracias al desarrollo de tratamientos paliativos en las últimas decadas, la esperanza de vida es ahora mucho mayor (en torno a 30-40 años). Estos tratamientos consisten en antibióticos para las infecciones respiratorias, fisioterapia respiratoria y ejercicio para mejorar la función pulmonar, una buena nutrición y suplementos de enzimas si hay insuficiencia pancreática.
Síntomas

- Retraso en el crecimiento
- Incapacidad para aumentar de peso normalmente durante la niñez
- Ausencia de deposiciones durante las primeras 24 a 48 horas de vida
- Piel con sabor salado
- Dolor abdominal a causa del estreñimiento grave
- Aumento de gases, meteorismo o un abdomen que parece hinchado (distendido)
- Náuseas e inapetencia
- Pérdida de peso
- Tos o aumento de la mucosidad en los senos paranasales o los pulmones

- Fatiga
- Congestión nasal causada por los pólipos nasales.
- Episodios recurrentes de neumonía
- Dolor o presión sinusal causados por infección

- Esterilidad (en los hombres)
- Inflamación repetitiva del páncreas
- Síntomas respiratorios
- Dedos malformados

Anemia Drepanocítica

También conocida como enfermedad de células falciformes (SCD, por sus siglas en inglés) es un grupo de trastornos hereditarios de los glóbulos rojos. Los glóbulos rojos saludables son redondos y se mueven a través de pequeños vasos sanguíneos para transportar oxígeno a todas las partes del cuerpo. En una persona que tiene SCD, los glóbulos rojos se endurecen, se vuelven pegajosos y tienen la apariencia de una herramienta agrícola en forma de C denominada “hoz”.
Las células falciformes o drepanocitos mueren anticipadamente, lo cual provoca una escasez constante de glóbulos rojos. Además, cuando los drepanocitos circulan a través de vasos sanguíneos pequeños, se atascan y obstruyen el flujo sanguíneo. Esto puede causar dolor y otros problemas graves como infecciones, síndrome torácico agudo y accidente cerebrovascular.
Causa de la enfermedad de células falciformes
La SCD es una afección genética que se presenta en el nacimiento. Es heredada cuando un niño recibe dos genes de drepanocitos, uno de cada padre.
Diagnóstico

La enfermedad de células falciformes se diagnostica con un análisis de sangre sencillo. Con mucha frecuencia se detecta en el nacimiento durante las pruebas de detección de rutina que les hacen a los recién nacidos en el hospital. Asimismo, la SCD puede diagnosticarse antes del nacimiento.
Debido a que los niños con SCD tienen un mayor riesgo de padecer infecciones y otros problemas de salud, el diagnóstico y el tratamiento precoces son importantes.
Puede llamar a su a su organización de la enfermedad de células falciformes para averiguar cómo puede hacerse los análisis.
Complicaciones y tratamientos

Las personas con SCD comienzan a presentar signos de la enfermedad durante el primer año de vida, normalmente alrededor de los 5 meses de edad. Los síntomas y las complicaciones de la SCD son diferentes para cada persona y pueden variar de leves a graves.
La única cura para la SCD es un trasplante de médula ósea o de células madre.
La médula ósea es un tejido blando y adiposo que se encuentra en el centro de los huesos, donde se forman las células sanguíneas. El trasplante de médula ósea o de células madre es un procedimiento en el que se obtienen células saludables que producen sangre de una persona, el donante, y se las trasplanta a otra persona cuya médula ósea no funciona adecuadamente.
Los trasplantes de médula ósea o de células madre son muy riesgosos y pueden tener efectos secundarios graves, incluso la muerte. Para que el trasplante funcione, la médula ósea debe tener una compatibilidad alta. Por lo general, el mejor donante es un hermano o una hermana. Los trasplantes de médula ósea o de células madres se realizan solo en casos de SCD grave en niños con daños mínimos en los órganos a causa de la enfermedad.
Fenilcetonuria

La fenilcetonuria es una enfermedad hereditaria, también llamada PKU, que consiste en una alteración del metabolismo en la que el cuerpo es incapaz de descomponer un aminoácido llamado fenilalanina, el cual se encuentra en la mayoría de los alimentos. Esto provoca que el organismo no pueda metabolizar en el hígado el aminoácido tirosina que se forma a partir de la fenilalanina.
Su herencia es autosómica recesiva, es decir, que debe heredarse la mutación del gen de la fenilalanina hidroxilasa de los dos progenitores. Esto sucede debido a que las personas con fenilcetonuria carecen de la enzima fenilalanina hidroxilasa (FAOH), lo que origina un aumento de la concentración sanguínea de fenilalanina, al impedir que esta se transforme a tirosina.

Además, como consecuencia de esta carencia, se activa una vía metabólica alternativa que produce una serie de componentes: fenilpiruvato, fenilactato y fenilacetato. Estos componentes son perjudiciales para el organismo y ocasionan daños en el sistema nervioso central y en el cerebro.
El pronóstico puede ser bueno si se detecta debidamente y de forma precoz y se instaura un tratamiento a tiempo, basado fundamentalmente en el seguimiento de una dieta estricta baja en fenilalanina en el primer año de vida del niño y durante su etapa de crecimiento, dieta que probablemente deberá seguirse toda la vida, con el fin de evitar o disminuir los daños mentales.
Causas

La causa fundamental para padecer fenilcetonuria (PKU) es el déficit de la fenilalanina hidroxilasa. Esto condiciona que se altere la conversión de fenilalanina en tirosina y por tanto se produzca un aumento en sangre de fenilalanina y de sus productos, fundamentalmente de fenilacetato y fenillactato. Los niveles de tirosina pueden permanecer normales o bajos en pacientes con PKU.
Las consecuencias de esta alteración metabólica son que los niveles elevados de fenilalanina van a interferir negativamente en el crecimiento y madurez del cerebro, en la producción de transmisores nerviosos y en la mielinización (sustancia fundamental para el correcto funcionamiento de las conexiones nerviosas).
El desarrollo neurológico y cerebral se ve interferido también por los niveles elevados de los metabolitos de la fenilalanina (fenilacetato y fenillactato). Todas estas sustancias por encima de los valores deseables condicionan una alteración del desarrollo intelectual.
Como el desarrollo intelectual y la madurez cerebral se producen a lo largo de la infancia, es en esta etapa y a consecuencia de las alteraciones descritas donde se apreciarán los síntomas de la fenilcetonuria.
Síntomas que presentan los bebés con fenilcetonuria:
- Retraso psicomotor.
- Cuadros psicóticos de tipo autista.
- Convulsiones.
- Síndrome de West.
- Eccema facial muy rebelde.
- Tamaño de la cabeza notablemente inferior a lo normal.
- Movimientos espasmódicos de brazos y piernas.
- Retraso mental.
- Temblores.
- Postura inusual de las manos.
Diagnóstico

Se realizan exámenes metabólicos de rutina a los neonatos para determinar si padecen fenilcetonuria, normalmente mediante una punción en el talón poco después del nacimiento.
Si da positivo hay que realizar exámenes de orina y de sangre para confirmar el diagnóstico de PKU. En ocasiones es posible que la prueba resulte negativa si se realiza antes de las 24 horas de vida. En estos casos, se puede recomendar repetir la prueba después de una o dos semanas de vida.
Se puede realizar la prueba del cloruro férrico en la orina. En casos excepcionales no se encuentra el ácido fenilpirúvico en la orina siempre, sino solo cuando se ha expuesto al niño a una sobrecarga dietética o cuando tiene fiebre. Estos casos se consideran como defectos parciales.
Otra prueba es la técnica de Guthrie, que consiste en la detección de la fenilalanina mediante la inhibición que produce el metabolito beta 2 tienialanina sobre el crecimiento del Bacillus subtilis. Este test tiene un porcentaje de sensibilidad y especificidad del 99%.
En casos conocidos de fenilcetonuria, se puede aconsejar el estudio genético a los familiares para detectar posibles portadores, mediante la detección de la mutación.
Enfermedad ligada al cromosoma X
Hemofilia

La Hemofilia es una enfermedad que afecta a la coagulación de la sangre ya que se caracteriza por un defecto en alguno de
factores que la componen, que se necesitan para que la sangre coagule.
La coagulación de
la sangre es como una fila de “fichas” de dominó que situamos una detrás de
otra. Al empujar la primera, ésta hace caer sucesivamente a todas las demás. La
Hemofilia sería esa situación pero una de las “fichas” intermedias es más
corta, defectuosa, que no es capaz de empujar a su siguiente en la fila con lo
que el orificio no se tapa y la sangre se pierde. Así es la cascada de la
coagulación en que las “fichas” son los factores y en que la última ficha es el
coagulo de fibrina.
Transmisión
hereditaria

La Hemofilia es
una enfermedad hereditaria. Su defecto se encuentra en el cromosoma X, es decir, el cromosoma que se relaciona con el
sexo trasmitida por las mujeres (portadoras) y la padecen
los hombres debido a la
dotación de dos cromosomas X (XX) de la mujer y una dotación XY en el hombre.
La transmisión de la Hemofilia se dice que es recesiva y no dominante ya que
puede que no aparezca en una generación siguiente (salto de generación) por la
simple razón de que se den portadoras sanas o varones sanos, y sí aparezca en otra generación posterior. Aquí,
hay una explicación más gráfica de la trasmisión de la Hemofilia.
Causas de la
hemofilia
La causa de que un
factor no funcione es, que el organismo lo sintetice defectuoso y como se trata
de una enfermedad hereditaria esto significa que el defecto se encuentra en una región del ADN (gen) que da lugar a una proteína que es el factor.
En el caso de la Hemofilia A el defecto más habitual es un gran cambio, llamado inversión del intrón 22, en el que para entendernos pondríamos las páginas de la segunda mitad
del libro al principio. Pero el defecto se puede deber también a pequeños
cambios de una letra (mutaciones puntuales) a eliminar algunas frases
(deleciones) o a meter frases o palabras al azar dentro de una página
(inserciones). En el caso de la Hemofilia B los errores se deben también a mutaciones puntuales, deleciones o inserciones pero también a la eliminación de unas cuantas páginas del libro o al intercambio
de páginas de un libro por las de otro que nada tiene que ver con el primero.
Sintomatología

La Hemofilia, en general, ya sea del tipo A o del tipo B, se
caracteriza por manifestaciones
hemorrágicas espontáneas o bien
por un sangrado excesivo cuando se produce algún tipo de traumatismo. Así, se deben distinguir las hemorragias articulares, las musculares y las de otra índole que, en ocasiones, pueden ser
graves.
La articulación se
torna rígida, dolorosa al moverla e inestable. Se vuelve todavía más inestable
a medida que los músculos que la rodean se debilitan. Estas hemorragias se
producen, fundamentalmente, en rodilla en un 44%, en codo en un 25%, en tobillo en un 15%, en hombro en un 8%, en cadera en un 5% y en otras
localizaciones en el 3% de
los casos.
Hoy día el
paciente con Hemofilia no muere de una simple o moderada ni siquiera grave
hemorragia si es tratado adecuadamente; el problema clínico sanitario es la
artropatía hemofílica.
Tratamiento

La Hemofilia, se
trata mediante la administración por vía
intravenosa del factor deficiente VIII o IX a la dosis adecuada en función de la edad y grado de severidad del episodio hemorrágico. Los concentrados utilizados de factores pueden
ser plasmáticos o recombinantes, ambos sometidos a procesos de inactivación viral.
A pesar de que este tratamiento es el adecuado algunos pacientes producen una
respuesta inmune contra el factor exógeno administrado, respuesta que será
tanto mayor cuanto mayor sea la porción de factor defectuosa. Estos pacientes
que desarrollan de esta manera los llamados “inhibidores”, se tratan con concentrados de mezclas de factores
de la coagulación o con Factor VII activado.
Daltonismo

El daltonismo (también
llamado deficiencia o ceguera de color),
ocurre cuando los colores no pueden ser vistos de manera normal. Comúnmente, el
daltonismo ocurre cuando alguien no puede distinguir entre ciertos colores, por
lo general entre verdes y rojos, y ocasionalmente azules.
En la retina (el tejido sensible a la luz que
recubre la parte posterior del ojo), hay dos tipos de células que detectan la
luz: los bastones y conos. Los bastones sólo detectan la luz y la oscuridad, y
son muy sensibles a bajos niveles de luz. Los conos detectan el color y se
concentran cerca del centro de su visión. Hay tres tipos de conos que ven el
color: rojo, verde y azul. El cerebro utiliza la información proveniente de
éstas tres células cónicas de color para determinar nuestra percepción del
color.
El daltonismo puede ocurrir cuando una o más
de las células cónicas de color están ausentes, no funcionan correctamente, o
detectan un color diferente al normal. Un daltonismo severo ocurre cuando hay
una ausencia de los tres conos; un daltonismo leve ocurre cuando hay los tres
conos están presentes, pero alguna de las células cónicas no funciona
normalmente y detecta un color diferente al normal.
Hay diferentes grados de daltonismo. Algunas
personas con deficiencias leves de color pueden ver los colores normalmente con
buena luz, pero tienen dificultades cuando la luz es tenue. Otras no pueden
distinguir ciertos colores en cualquier tipo de luz. La forma más grave de
daltonismo, en el cuál todo se ve en tonos grises, es poco común. El daltonismo
usualmente afecta a ambos ojos por igual y se mantiene estable a lo largo de la
vida.
Causas de daltonismo

La mayoría de las personas con daltonismo nacen con
la condición. (Esto se denomina enfermedad
congénita.) Los defectos congénitos de la visión del color
usualmente se transmiten de madres a hijos varones.
Dichos defectos se deben a la falta parcial o total
de foto receptores sensibles a la luz (conos) en la retina (la capa de células
nerviosas sensibles a la luz que recubren la parte posterior del ojo). Los
conos ayudan a distinguir los colores rojo, verde y azul.
La mayoría de los problemas de visión de color
ocurren más tarde durante el transcurso de la vida, como resultado de
enfermedades, trauma, efectos tóxicos de drogas, y enfermedades metabólicas o
vasculares. Los defectos de la visión de color provenientes de alguna
enfermedad, son poco entendidos en comparación con los problemas congénitos. Un
daltonismo causado por alguna enfermedad afecta a los dos ojos en forma
diferente, y por lo general empeora con el tiempo. Una pérdida de la visión de
color adquirida puede ser el resultado de daños en retina o en el nervio
óptico.
Síntomas de daltonismo

Los síntomas de daltonismo pueden variar de leves a
severos. Muchas personas tienen síntomas tan leves que no son conscientes de
que tienen una deficiencia de color. Los padres sólo pueden notar un problema
en su hijo cuando está aprendiendo los colores. Los síntomas incluyen:
- Dificultad para ver los
colores y su brillo en forma usual;
- Incapacidad de establecer
una diferencia entre los diferentes tonos de un color u otros similares,
especialmente el color rojo y el verde, o el azul y el amarillo.
A excepción de la forma más grave de daltonismo, la
condición no afecta la nitidez de la visión. La incapacidad absoluta de ver
cualquier color, solamente tonos grises, es llamada acromatopsia. Ésta
condición es rara y a menudo es asociada con la ambliopía, el nistagmo (movimiento
involuntario y rápido de los ojos), sensibilidad a la luz, y una baja visión.
Diagnóstico y tratamiento del daltonismo

Se lleva a cabo una sencilla prueba para determinar
si se tiene daltonismo, la prueba consiste en mostrarle un patrón compuesto de
múltiples puntos de color. Si usted no tiene una deficiencia de color, podrá
identificar los números y formas entre los puntos. Si usted es daltónico, le
será difícil encontrar los números y formas dentro del patrón, o simplemente
puede no encontrarlos.
No existe un tratamiento para el daltonismo
congénito. Por lo general, el daltonismo no causa una discapacidad importante.
Sin embargo, hay lentes de contacto y anteojos especiales que pueden ayudar a
las personas con daltonismo a distinguir diferencias entre colores similares.
Las formas adquiridas de daltonismo pueden ser
tratadas identificando la enfermedad o droga que lo causa.
Enfermedades infecciosas
Endocarditis
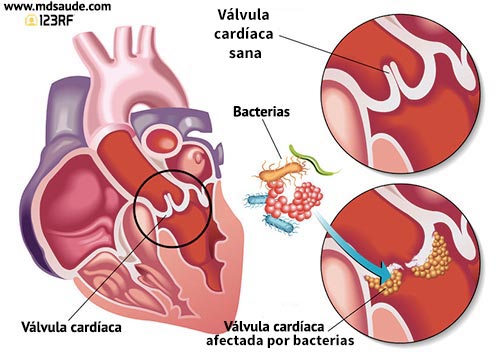
Es una inflamación del revestimiento interno de las cámaras y válvulas
cardíacas (endocardio). Es causada por una infección bacteriana o, en raras
ocasiones, fúngica.
Causas

La endocarditis puede comprometer el miocardio, las válvulas o el
revestimiento del corazón. Algunas personas que se enferman de endocarditis
tienen:
·
Una anomalía congénita del corazón
·
Una válvula
cardíaca dañada o anormal
·
Antecedentes de
endocarditis
·
Una válvula cardíaca
nueva después de cirugía
La endocarditis se inicia con la entrada de gérmenes en el torrente
sanguíneo que luego viajan hasta el corazón.
·
La infección
bacteriana es la causa más común de endocarditis.
·
La endocarditis
también puede ser causada por hongos, tales como cándida.
·
En algunos casos,
no se puede encontrar la causa.
Los gérmenes tienen más probabilidades de entrar en el torrente
sanguíneo durante:
·
Catéteres venosos centrales
·
Uso de drogas
inyectadas, por la utilización de agujas sucias (sin esterilizar)
·
Cirugía dental
reciente
·
Otras cirugías o
procedimientos menores en las vías respiratorias, las vías urinarias, piel
infectada, o huesos y músculos
Síntomas

Los síntomas de endocarditis se pueden desarrollar de forma lenta o
repentina.
La fiebre, los escalofríos y la sudoración son síntomas frecuentes.
Estos algunas veces pueden:
·
Estar presentes
durante días antes de que aparezca cualquier otro síntoma
·
Aparecer y
desaparecer o ser más notorios durante la noche
Usted también puede presentar fatiga, debilidad y dolores en los
músculos o articulaciones.
Otros signos incluyen:
·
Pequeñas zonas de
sangrado bajo las uñas (hemorragia lineal subungueal)
·
Manchas cutáneas
(piel) rojas e indoloras en las palmas de las manos y en las plantas de los
pies (lesiones de Janeway)
·
Ganglios rojos y
dolorosos en las yemas de los dedos de la manos y de los pies (nódulos de
Osler)
·
Dificultad para respirar con la actividad
·
Inflamación de
pies, piernas y abdomen
Pruebas y exámenes
El proveedor de atención médica puede detectar un
nuevo soplo cardíaco o un cambio en un soplo cardíaco previo.
El examen oftalmológico puede mostrar sangrado en la retina con una zona
central de aclaramiento. Este hallazgo se conoce como manchas de Roth. Puede
haber pequeñas áreas de sangrado en la superficie del ojo o los párpados.
Los exámenes que se pueden hacer incluyen:
·
Hemocultivo para ayudar a identificar la bacteria o el hongo que está causando la
infección.
·
Conteo sanguíneo completo (CSC), proteína C reactiva (PCR) o tasa de sedimentación eritrocítica (ESR,
por sus siglas en inglés).
·
Una ecocardiografía de rutina o una ecocardiografía transesofágica para observar las válvulas cardíacas.
Tratamiento

Usted puede necesitar estar en el hospital para recibir antibióticos por
vía intravenosa. Los hemocultivos y los exámenes de sangre ayudarán al
proveedor de atención a escoger el mejor antibiótico.
Luego, se necesitará una terapia de antibióticos a largo plazo.
·
En la mayoría de
los casos, la gente necesita terapia durante 4 a 6 semanas para eliminar por completo
todas las bacterias de las cámaras y válvulas del corazón.
·
Los tratamientos
con antibióticos que se inician en el hospital se deberán continuar en
casa.
Se suele necesitar cirugía para reemplazar la válvula cardíaca cuando:
·
La infección se
está separando en pequeños fragmentos, lo cual ocasiona una serie de accidentes
cerebrovasculares.
·
La persona
presenta insuficiencia cardíaca como resultado de los daños a las válvulas del
corazón.
Meningitis

La meningitis es una enfermedad infecciosa provocada por virus o bacterias que en muchas ocasiones se encuentran
en la nariz y en la garganta de personas sanas (portadores sanos) que la
contagian a terceras personas.
La
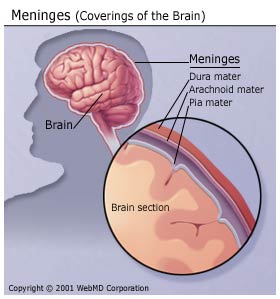
enfermedad provoca la infección e inflamación de las
meninges, unas membranas que rodean el cerebro y la médula
espinal. Cuando se infectan sólo las meninges se habla de meningitis. En
algunas ocasiones, la bacteria se introduce en la sangre diseminándose. A este
problema se le denomina sepsis meningocócica. También puede ocurrir que se den
ambos problemas a la vez.
Causas

Las
causas más frecuentes de la meningitis son las infecciones, que pueden estar provocadas por virus o por
bacterias.
La más común, y menos grave, es la infección vírica, que
generalmente mejora sin que se la administre ningún medicamento al paciente.
Sin embargo, las bacterianas son muy graves y pueden llegar a provocar daños
cerebrales e incluso la muerte.
Además
de por las infecciones, la meningitis puede aparecer debido a hongos,
tumores, y otros virus como el de las paperas,
el VIH,
el herpes labial y
el genital.
Contagio
El
contagio de esta patología se produce a través de la saliva y las gotitas que
se expulsan al hablar, estornudar o toser. El contagio a través de objetos no
es habitual y
ocurre en pocas ocasiones.
La
meningitis suele aparecer en otoño y en primavera. En entornos como las
guarderías, las escuelas o las residencias puede propagarse con rapidez.
Síntomas

Una
vez contagiada, una persona puede tener los primeros síntomas entre los dos y
los diez días posteriores al contagio. En ocasiones tiene un comienzo brusco
con síntomas similares a los de un catarro o una gripe.
Los más frecuentes y que anuncian la gravedad son:
·
Fiebre alta.
·
Dolor de cabeza intenso.
·
Rigidez de la nuca.
·
Vómitos bruscos.
·
Somnolencia.
·
Pérdida de conciencia.
·
Agitación, delirio y/o convulsiones.
·
Manchas de color rojo-púrpura en la piel,
lo que implica una mayor gravedad.
Prevención
La mejor prevención es la vacunación.
Hasta hace poco sólo existían vacunas contra el Haemophilus tipo b, el
meningococo tipo A y C y el neumococo, que habían hecho prácticamente
desaparecer estos tipos de meningitis en nuestro entorno.
La
vacunación está indicada en los menores de 6 años y puede provocar efectos
secundarios leves, como inflamación y molestias en el lugar de la inyección,
fiebre baja e irritabilidad.
Está contraindicada cuando existen estados
febriles en el momento de la vacunación; si el niño es hipersensible a alguno
de los componentes de la vacuna, o en personas inmunodeprimidas.
Las
dosis varían en función de la edad. Si se inicia la vacunación entre los 2 y
los 5 meses se administrarán 4 dosis; de 6 a 23 meses 3 dosis; de 2 a 10 años 2
dosis; adolescentes y adultos 2 dosis.
Diagnóstico
El
diagnóstico de la meningitis bacteriana se realiza analizando una muestra de líquido cefalorraquídeo (de la médula espinal). Dicha muestra
se obtiene mediante una punción en la columna vertebral (punción lumbar).
Además,
el especialista puede pedir otras pruebas para confirmarlo, como una ecografía o
una tomografía axial computadorizada (TC)
que permitan determinar si existe un absceso responsable de la meningitis.
Tratamientos
La
mayor parte de las personas que sufre una meningitis viral se cura sin
problemas.
En
la meningitis bacteriana el tratamiento consiste en cuidados específicos en el hospital y terapia intensa con antibióticos.
Otros
cuidados que se le pueden prescribir al paciente son la administración de
líquidos por vía intravenosa y medicamentos para tratar lesiones asociadas que
pueden aparecer, como el edema cerebral, el shock o las crisis epilépticas.
Es
imprescindible el diagnóstico precoz y la rápida asistencia del
especialista. En algunos casos la enfermedad evoluciona con
gran fuerza o afecta a personas con el sistema inmune débil y pueden provocar
desenlaces fatales.
Septicemia

La
septicemia, también conocida como bacteriemia, se produce cuando una infección
bacteriana ingresa en el torrente sanguíneo. Si no se trata, puede avanzar
rápidamente y convertirse en una septicemia grave, una complicación importante
de una infección que se caracteriza por una inflamación generalizada en el
organismo. Esta inflamación puede causar la formación de coágulos sanguíneos e
impedir que el oxígeno llegue a los órganos vitales, lo que podría dar lugar a
una insuficiencia orgánica y, en algunos casos, la muerte.
Causas
La
causa fundamental de la septicemia es una infección bacteriana (generalmente
grave) en otra parte del organismo. Las infecciones en las vías urinarias, los
pulmones y el área abdominal son causas posibles de septicemia. Las bacterias
responsables de estas infecciones ingresan en el torrente sanguíneo y se
multiplican, por lo cual los síntomas se manifiestan de inmediato.
Síntomas

Si no
se trata la infección en la sangre, los síntomas de la septicemia pueden
avanzar y convertirse rápidamente en una afección más grave. Incluso en las
primeras etapas de la enfermedad, una persona que tiene septicemia luce muy
enferma.
Entre
los primeros síntomas, generalmente se incluyen los siguientes:
·
escalofríos
·
temperatura corporal alta
·
respiración o frecuencia cardíaca muy
aceleradas
Entre
los demás síntomas que suelen manifestarse a medida que la septicemia avanza,
se incluyen los siguientes:
·
confusión o incapacidad para pensar
con claridad
·
aparición de manchas rojas en la piel
·
reducción en el volumen de orina
·
flujo sanguíneo anormal (choque)
Complicaciones
Choque
septicémico
Una de
las complicaciones de la septicemia es una disminución extrema de la presión
arterial que recibe el nombre de choque septicémico. Las toxinas liberadas por
las bacterias en el torrente sanguíneo pueden reducir drásticamente el flujo
sanguíneo, lo que puede provocar daños en los órganos o los tejidos. El choque
septicémico es una emergencia médica y quienes lo sufren generalmente reciben
atención en la unidad de cuidados intensivos de un hospital. Es posible que los
pacientes que sufren un choque septicémico deban ser conectados a un
respirador. La tasa de mortalidad de una septicemia que deriva en un choque
septicémico es del 50 por ciento (Johns Hopkins).
Diagnóstico

Para
diagnosticar la septicemia, el médico evaluará los síntomas y los antecedentes
médicos del paciente antes de realizar una exploración física a fin de detectar
una disminución en la presión arterial o la temperatura corporal. Además, el
médico puede buscar signos de afecciones que suelen aparecer junto con la
septicemia. Es posible que le realicen un urocultivo y un hemocultivo y que le
tomen muestras de las llagas que pueda haber en la piel para confirmar la
presencia de una infección bacteriana.
Entre
las pruebas que pueden realizarse, se incluyen las siguientes:
·
recuentos de células sanguíneas y
plaquetas
·
pruebas para evaluar la coagulación
sanguínea
·
pruebas para medir los niveles de
oxígeno y dióxido de carbono en la sangre (si la septicemia provoca problemas
respiratorios)
Tratamiento

La
septicemia es una emergencia médica que debe tratarse en el hospital. Muchas
personas que tienen septicemia son internadas en la unidad de cuidados
intensivos (UCI) de un hospital para su tratamiento y recuperación. El
tratamiento depende de distintos factores, incluidos los siguientes:
·
la edad y el estado de salud general
del paciente, la gravedad de la afección, la tolerancia a determinados
medicamentos. Se
usarán antibióticos para tratar la infección bacteriana responsable de la
septicemia.
Prevención
Las
infecciones bacterianas son la causa subyacente de la septicemia. Por ende, una
de las mejores maneras de prevenirla es consultar al médico de inmediato si
considera que podría presentar una infección. Si la infección puede tratarse
eficazmente con antibióticos en sus primeras etapas, es probable que logre
impedir que las bacterias ingresen en el torrente sanguíneo. Los padres pueden
proteger a sus hijos contra la septicemia si se aseguran de que reciban todas
las vacunas que les corresponden.
